
U.S. FDA 510(k)
“All information in this page are from official U.S. FDA. website.”
510(k) Submission Process
Introduction
Premarket Notification (510(k)) submissions for medical devices are reviewed by FDA’s Center for Devices and Raidological Health (CDRH), specifically, by the Office of Device Evaluation (ODE) and the Office of In Vitro Diagnostics and Raidological Health (OIR). The Divisions in these offices are organized according to medical device specialties. 510(k) submissions are reviewed by ODE and OIR staff, including biomedical engineers, physicians, microbiologists, chemists, and other scientific professionals.
Log-in and Acknowledgement Procedure
A 510(k) submitter should submit two copies of its 510(k) to CDRH’s Document Control Center (DCC) at the following address:
Food and Drug Administration
Center for Devices and Raidological Health
Document Control Center – WO66-G609
10903 New Hampshire Avenue
Silver Spring, Maryland 20993-0002
One of the two copies must be an electronic copy (an eCopy).
When the DCC receives the 510(k) submission, it assigns the submission a unique control number. This number is commonly referred to as the “510(k) number,” or “K number.” The 510(k) number begins with the letter “K” followed by 6 digits. The first two digits designate the calendar year the submission was received; the last four digits represent the submission number for the year, starting with 0001 and increasing with each new submission.
For example: the first 510(k) submission for the calendar year 2013 would be K130001.
The DCC then conducts two verification checks to confirm that:
The proper user fee payment was received for the submission.
Note: the user fee amount to be paid is based on when the 510(k) is received by FDA and not the date on which it was sent by the submitter.
A valid eCopy of the 510(k) submission was provided.
If the proper user fee has not been paid and/or a valid eCopy has not been provided, then the DCC will email a Hold Letter to the 510(k) submitter, usually within 7 days of receipt of the 510(k). The submitter then has 180 calendar days from the date of the Hold Letter to fully resolve the issues with the user fee or eCopy submission. If the issues are not resolved within 180 days, then the 510(k) is considered to be withdrawn and deleted from our review system, and the submitter will need to submit a new, complete 510(k) to pursue FDA marketing clearance.
If the proper user fee has been paid AND a valid eCopy has been provided, the DCC will email an Acknowledgment Letter to the contact person identified in the 510(k) submission. The Acknowledgement Letter identifies:
the date of receipt (this is the date that FDA received the 510(k) submission, the proper user fee payment, and valid eCopy); and
the 510(k) number assigned to the submission.
Note: The Acknowledgment Letter is NOT a marketing clearance letter. The 510(k) number identified in the Acknowledgement Letter should be referenced in all further correspondence with FDA regarding the 510(k). Failure to reference the 510(k) number may result in processing delays.
Acceptance Review
After the Acknowledgement Letter is sent, the DCC routes the 510(k) to the appropriate ODE or OIR Division, based on the device type and medical specialty that is listed in the 510(k) submission.
Upon receipt in the Division, the 510(k) is assigned to the appropriate Branch, and then assigned to a Lead Reviewer. The Lead Reviewer conducts the Acceptance Review using the appropriate Acceptance Checklist in FDA’s Guidance titled Refuse to Accept Policy for 510(k)s. In the Acceptance Review, the Lead Reviewer determines whether the 510(k) submission meets the minimum threshold of acceptability and should be accepted for substantive review.
Within 15 days of the receipt of the submission, the submitter will receive an electronic notification of the Acceptance Review result, which will:
identify the name and contact information of the FDA Lead Reviewer assigned to the 510(k); and
indicate the status of the 510(k).
The Acceptance Review result will be one of the following:
the 510(k) was accepted for substantive review; or
the 510(k) was not accepted for review (i.e., considered refused to accept or RTA); or
the 510(k) is under substantive review because FDA did not complete the acceptance review within 15 calendar days.
A 510(k) not accepted for review is placed on RTA Hold. The submitter has 180 calendar days to fully address the deficiencies cited in the RTA Hold. If this is not done, the 510(k) is considered withdrawn and deleted from our review system. If the 510(k) is deleted, the 510(k) submitter will need to submit a new, complete 510(k) to pursue FDA marketing clearance for that device.
Once accepted, a 510(k) proceeds to the Substantive Review.
Substantive Review (including Substantive Interaction and Interactive Review)
During Substantive Review, the Lead Reviewer conducts a comprehensive review of the 510(k) submission and communicates with the submitter through a Substantive Interaction, which should occur within 60 calendar days of receipt of the 510(k) submission
The Substantive Interaction communication is typically:
an email stating that FDA will proceed to resolve any outstanding deficiencies via Interactive Review; or
an Additional Information (AI) request which places the submission on hold.
Interactive Review
If the Lead Reviewer chooses to continue with an Interactive Review, this means the Lead Reviewer has determined that any outstanding deficiencies may be adequately addressed within the timeframe set by the Medical Device User Fee Amendment of 2012 (MDUFA III) performance goal for a 510(k) (90 FDA days) and that the submission will not be placed on hold. The Lead Reviewer communicates with the submitter during the Interactive Review using tools such as:
Email
Telephone Call
During Interactive Review, the Lead Reviewer may request additional information from the submitter, who may either send the information to the Lead Reviewer directly or to the DCC.
Note: During Interactive Review, any information submitted to the DCC must include a valid eCopy.
Additional Information (AI) Request
If the Lead Reviewer sends an AI Request, the submission is placed on hold. The submitter has 180 calendar days from the date of the AI Request to submit a complete response to the AI Request. Note: The response must be received in the DCC within 180 calendar days of the date of the AI Request. No extensions beyond 180 days are granted. If FDA does not receive a complete response to all deficiencies in the AI Request within 180 days of the date of the AI Request, the submission will be considered withdrawn and deleted from our review system. If the 510(k) is deleted, the 510(k) submitter will need to submit a new 510(k) to pursue FDA marketing clearance for that device.
The submitter must submit the response, with a valid eCopy, to the DCC. The response should:
include the submitter’s name;
list the 510(k) number;
identify the submission as Additional Information (AI) to the 510(k);
list the date of FDA’s request for additional information; and
provide the requested information in an organized manner.
For more information on Substantive Review and Interactive Review, please see FDA’s Guidances:
FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals and
Types of Communication During the Review of Medical Device Submissions.
510(k) Decision Letter
The FDA goal to make a MDUFA Decision for a 510(k) is 90 FDA Days. FDA Days are calculated as the number of calendar days between the date the 510(k) was received and the date of a MDUFA decision, excluding the days the submission was on hold for an AI request. MDUFA Decisions for 510(k) submissions include findings of substantially equivalent (SE) or not substantially equivalent (NSE).
When a decision is made, FDA will issue the decision letter to the submitter by email to the email address provided in the 510(k) cover letter.
A 510(k) that receives an SE decision is considered “cleared.” FDA adds the cleared 510(k) to the 510(k) database, which is updated weekly. The IFU and the summary will be sent as attachments to the SE letter. The IFU will not be signed since it is considered an attachment to the SE letter. Therefore, the signature on the SE letter will apply to both the letter and the IFU.
If FDA does not reach a MDUFA decision within 100 FDA days (i.e., 10 days after the MDUFA goal), FDA will issue a Missed MDUFA Communication, which is written feedback to the submitter to be discussed in a meeting or teleconference, including the major outstanding review topic areas or other reasons that are preventing FDA from reaching a final decision, with an estimated date of completion.
Timeline of Communication with 510(k) Applicants
FDA follows the MDUFA III performance goals for review of 510(k) submissions. The following flow chart provides a simplified summary of event and interaction milestones during the course of a 510(k) submission. This flow chart is based on the goals and procedures outlined in the MDUFA III Performance Goals and Procedures Commitment Letter. For more information please see MDUFA III Performance Goals and Procedures.
Timeline of Communication during 510(k) Review
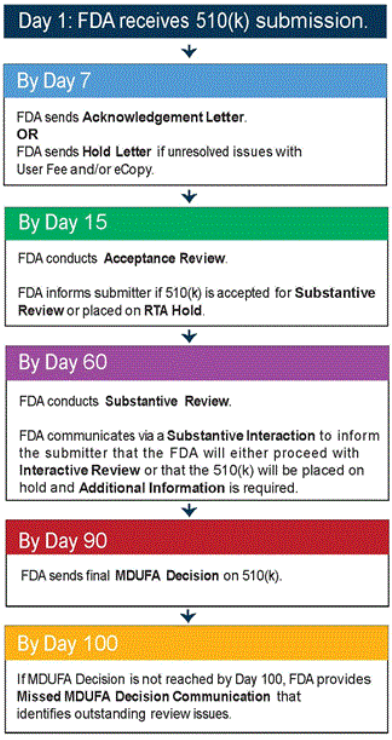
Days are Calendar Days.
The timeline is based on the performance goals set by Medical Device User Fee Amendments of 2012 (MDUFA III).
This timeline has been simplified.
Please Contact us for any further question.