국내 의료기기 인허가
IX. 식품의약품안전처 (MFDS) 의료기기 GMP
(의료기기 제조 및 품질관리기준) 심사
하기의 정보는 『의료기기법』(법률 제13116호), 『의료기기법시행령』(대통령령 제263375호), 『의료기기법시행규칙』(총리령 제1181호), 『의료기기 제조 및 품질관리기준』 (고시 제2015-71호)에 의거하여 작성되었다.
- GMP 심사의 목적
의료기기 시행규칙 6조/8조/26조/27조/30조/31조에 의거하여 제조허가 또는 제조인증을 받거나 제조신고를 하려는자는 총리령으로 정하는 바에 따라 필요한 시설과제조 및 품질관리체계를 미리 갖추어 허가 또는 인증을 신청하여야 한다.
- 적용 대상
의료기기 제조업자
의료기기 수입업자
임상시험용 의료기기 제조/수입자
- 적용 제외 대상
1등급 의료기기
수출만을 목적으로 제조하는 의료기기
- 심사종류
최초심사: 제조 또는 수입 의료기기가 기준에 적합함을 인정받기 위해 최초로 받아야 하는 심사
정기심사: 최초심사 후 3년 주기로 실시하는 사후 관리 심사
추가심사: 품목군을 새로이 추가하는 심사
변경심사: 제조소의 소재지 변경에 따라 적합성평가를 새로이 받아야 하는 심사.
- 의료기기 GMP 적합성평가 품목군 및 등급 분류
「의료기기 제조 및 품질관리기준」(식약처고시) 별표3의 26개 품목군과 의료기기 등급(1~4등급)으로 구분하여 적합성평가를 받도록 한다.
- GMP 적합성평가 절차
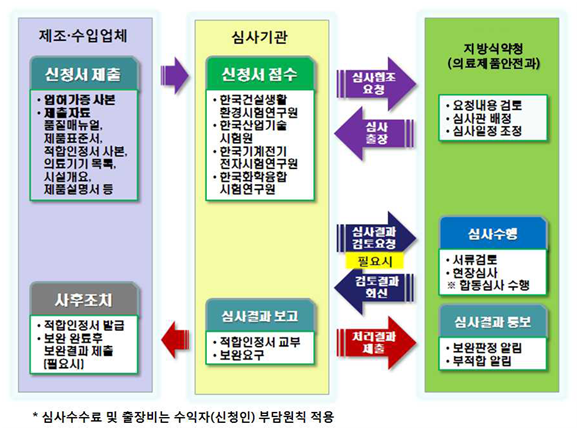
<출처: 의료기기 제조(수입) 허가•신고•심사 등 민원처리 안내서>>
- 구비서류
① 의료기기 품질관리기준 적합인정 신청서③④⑤
② 의료기기 제조(수입)업 허가증 사본(최초 신청일 경우 제외)
③ 다음 각 목의 적합성평가에 필요한 자료
- 제조소 개요(제조소의 명칭 및 주소, 제조소가 다수인 경우 모든 제조소명을 포함)
- 총 종업원의 수(신청 품목 품질에 영향을 줄 수 있는 모든 관련 인원)
- 해당 제조소에서 제조되는 의료기기 목록(품목명, 등급을 포함)
- 품질경영시스템 적합성 인증서 사본 (해당하는 경우)
- 평가 대상이 되는 각 제조소의 시설개요(평면도, 시설•장비 목록을 포함)
- 주요 공급업체의 소재지 및 업무범위(위탁공정 계약 등을 포함)
- 다른 인증기관으로부터 받은 실사결과 자료(해당되는 경우)
- 품질매뉴얼(품질 방침 포함, 수입의 경우 해외 제조소의 문서)
- 제품표준서(멸균, 소프트웨어 등 특수 공정 내용에 대한 설명 포함)
- 제품 설명서
- 사업자 등록증 사본
※ 「의료기기 제조 및 품질관리 기준」제7조제2항에 따라 구비서류 중 일부자료만 제출할 수 있음 제조소 정보(명칭, 주소, 약도, 주소지가 다수일 경우 모든 정보)
- 허가 기관 및 처리 기간
허가 기관: 의료기기품질관리심사기관
처리 기관: 신청접수일로부터 30일 내외
국내 의료기기 허가 관련 문의사항은 연락처의 정보로 문의 주시기 바랍니다.