
U.S. FDA 510(k)
510(k) 제출 절차
서론
시판 전 신고(510(k))를 위하여 제출된 문서는 미 식약청의 CDRH(Center for Device and Radiological Health: 의료기기 및 방사선 건강센터)의 부서들 중 생체공학자, 내과의, 미생물학자, 화학자 등과 의료기기 전문인들로 구성되어 있는 의료기기 평가 분과(ODE: Office of Device Evaluation)와 체외진단기 및 방사선 건강 분과(OIR: Office of In Vitro Diagnostics and Radiological Health)가 검토를 하게 된다.
접속 및 접수 통지 절차
510(k) 접수자는 아래의 CDRH의 DCC(Document Control Center: 문서관리과)의 주소로 두 부의 문서를 제출 하여야 한다:
Food and Drug Administration Center for Devices and Radiological Health Document Control Center – WO66-G609 10903 New Hampshire Avenue Silver Spring, Maryland 20993-0002
문서 두 부 중 한 부는 전자본으로 제출 되어야 한다.
문서관리과(DCC)에서 510(k) 제출 자료를 접수하면 해당 자료에 고유 번호를 부여하는데 일반적으로 이 번호를 “510(k)번호” 혹은 “K번호”라 칭한다. 이 K번호는 알파벳 K 뒤에 여섯 개의 숫자가 붙게 된다. 첫 두 숫자는 제출된 자료가 접수된 연도를 의미하며 뒤의 네 자리는 해당 연도에 몇 번째로 제출이 이루어 졌는지를 나타낸다. 즉, 해당 연도의 첫 제출자는 0001번이 부여되며 뒤로 갈수록 높은 숫자가 부여된다.
예) 2013년도에 최초로 접수 된 510(k) 자료의 번호: K130001
접수 후 문서관리과는 문서 접수 시 아래의 두 가지 사항을 확인한다.
- 적용되는 신청 비용의 접수 여부
참고: 510(k)의 제출 비용은 제출자가 문서를 발송 한 날짜가 아닌 미 식약청이 해당 문서를 접수한 날짜를 기준으로 부과된다. - 전자본이 접수 되었는지 확인
만일 합당한 수익자 부담금이나 전자본의 접수에 문제가 있을 경우 문서관리과에서는 통상적으로 문서 접수 7일 이내에 해당 510(k) 제출자에게 보류 통보를 보낸다. 제출자는 보류 통보의 발송일로부터 180일 이내에 수익자 부담금이나 전자본 접수에 관련된 문제를 해결해야 하며 180일 이내에 해결하지 못 할 경우 해당 510(k) 제출은 무효화 되며 전산에서 삭제된다. 추후 제출자는 새로운 신청절차를 통하여 재 신청 해야 한다.
합당한 수익자 부담금과 전자본의 접수가 정상적으로 이루어진 경우 문서관리과에서는 510(k) 제출 자료에 연락책으로 기재되어 있는 이메일로 접수확인서를 발송한다. 접수확인서에는 다음의 내용을 표기된다.
접수 일자 (수익자 부담금과, 전자본을 포함한 510(k) 문서 두 부가 모두 접수 완료 된 시점)
해당 제출에 부여되는 510(k) 번호
비고: 접수확인서는 시판 승인서가 아니다. 접수확인서에 기재되어 있는 510(k)번호는 해당 510(k) 승인 절차간 미 식약청과 이루어지는 모든 대응에 참조 되어야 한다. 510(k)번호가 참조되지 않은 대응은 진행의 지연을 초래할 수 있다.
인수 허가 검토
접수확인서의 발송 이후 문서관리부는 제출된 510(k) 자료를 기반으로 의료기기의 유형과 해당 전문 분야를 고려하여 ODE(Office of Device Evaluation: 의료기기 평가 분과) 혹은 OIR (Office of In Vitro Diagnostics and Radiological Health: 체외진단기 및 방사선 건강 분과)로 문서를 배정한다.
해당 문서를 접수한 분과는 적절한 부서를 배정하고 그 안에서 선임 검토자가 배정이 된다. 선임 검토자는 “510(k) 인수 불가 방침”이라는 미 식약청의 가이드문서에 포함되어 있는 적합한 인수 허가 체크리스트를 사용하여 인수 허가 검토를 실시하게 된다. 인수 허가 검토간 선임 검토자는 해당 510(k)제출 자료가 인수 허가의 최소 요구조건을 만족시키는지의 여부와 실질 검토를 실시하기 적합한지의 여부를 판단한다.
제출 15일 이내에 제출자는 인수 허가 검토 결과에 대하여 이메일 통지서를 접수하게 되는데 해당 통지서에는 아래의 사항들이 기재되어 있다.
해당 510(k) 제출에 배정된 선임 검토자의 이름과 연락처
해당 510(k) 제출의 현재 상황
인수 허가 검토의 결과는 아래 중 하나로 결정이 된다.
해당 510(k) 제출의 실질 검토가 승인 되었습니다.
해당 510(k) 제출은 검토가 승인되지 않았습니다. 혹은
미 식약청에서 해당 510(k) 제출에 대한 인수 허가 검토를 15일 내로 완료 하지 못하였음으로 실질 검토가 실시 실시되었습니다.
인수 불허가 결정된 510(k) 제출은 접수 거부의 보류상태로 남게 된다. 제출자는 접수 거부 사유에 대한 보완을 180일 내로 완료 해야 한다. 만일 기간 내에 적합한 조치가 취해지지 않을 경우 해당 510(k) 제출은 취소되며 시스템상에서 삭제된다. 삭제된 510(k)는 추후에 새로운 신청 절차를 통해 접수되어야 한다.
인수 허가가 결정된 510(k) 제출은 실질적 검토의 절차로 들어가게 된다.
실질 검토 (실질적 대화와 상호 검토)
실질적 검토 동안 선임 검토자는 해당 510(k) 제출에 대한 포괄적인 검토를 실시하며 실질적 대화 절차를 통하여 제출자와 소통하게 된다. 이러한 실질적 대화는 해당 510(k)가 접수 된지 60일 내에 이루어져야 한다.
실질적 대화는 일반적으로 아래의 사항들이 주를 이룬다.
미 식약청이 실질적 상호 보완을 통하여 존재하는 모든 부적합 사항을 해결할 것임을 표현한 이메일 혹은
추가적 정보 요청 (이 경우 해당 제출은 보류의 상태로 전환된다.)
상호 검토
만일 선임 검토자가 상호 검토의 절차를 지속하고자 선택했다면 이는 선임 검토자가 존재하는 모든 부적합 사항들을 2012년도 의료기기 수익자 부담금의 개정안 (MDUFA III: Medical Device User Fee Amendments III)에서 정한 기간(90일) 내에 해결이 가능하리라 판단했다는 의미이며 해당 제출 문서는 보류상태로 전환되지 않는다. 선임 검토자는 아래의 방식으로 제출자와 소통하게 된다.
이메일
전화
상호 검토의 절차간 선임 검토자는 제출자에게 추가 정보를 요청 할 수 있는데 제출자는 요청 자료를 선임 검토자에게 직접 보내거나 문서관리부로 보낼 수 있다.
비고: 상호 검토의 절차간 문서관리부로 접수되는 모든 자료는 유효한 전자본을 포함해야 한다.
추가 정보 요청 (AI: Additional Information Request)
일단 선임검토자가 추가 정보 요청을 할 경우 해당 제출은 보류 상태로 전환된다. 제출자는 해당 요청에 대한 답변과 자료를 완벽히 구비하여 180일 이내에 제출해야 한다. 비고: 요청에 대한 답변과 자료는 해당 요청이 이루어진 일자로부터 180일 이내에 미 식약처의 문서관리부에 접수가 완료 되어야 한다. 180일 이상의 연장은 불가하다. 만일 180일 이내에 모든 답변과 자료가 접수되지 않으면 미 식약처는 해당 510(k) 제출이 취소된 것으로 간주하고 시스템에서 삭제한다. 삭제된 제출은 새로운 신청 절 차에 의해서만 다시 접수 될 수 있다.
제출자는 답변과 자료를 보낼 때 반드시 유효한 전자본과 함께 제출해야 한다. 답변은 아래의 사항을 충족시켜야 한다.
제출자 성명
부여된 510(k) 번호
추가 정보 요청이 이루어진 날짜
요청 받은 정보와 자료
실질적 상호 보완과 상호 검토에 대한 상세 내용은 아래 미 식약청의 가이드 문서를 참고하면 된다.
시판전 신고 (510(k))시 미 식약청과 산업체의 행동강령: 미 식약처의 검토 기간과 목표
검토간 발생 할 수 있는 의사소통의 방식
510(k) 확정서
510(k) 절차에 대한 미 식약청의 공식 목표 기한은 90일이다. 90일의 측정은 추가 정보 요청이 발생하여 해당 제출이 보류의 상태에 놓인 기간을 제외하고 미 식약청에서 제출된 문서를 접수한 일자로부터 최종적인 결정까지 소요된 일자로 계산된다. 최종적인 결정은 본질적 동등성의 확인(SE: Substantially equivalent) 혹은 본질적으로 동등하지 않음(NSE: Not substantially equivalent)일 수 있다.
최종적인 결정이 이루어지면 미 식약청은 결정서를 발행하며 제출 자료에 기재되어 있는 이메일로 해당 결정서를 발송한다.
본질적으로 동등함으로 결정된 510(k) 제출은 “승인”된 것으로 여긴다. 승인된 510(k)는 매주 갱신되는 미 식약청의 510(k) 데이터베이스에 등록된다. 사용 설명서와 요약된 검토 결과가 본질적 동등성 승인서와 함께 제출자에게 송부된다. 첨부된 사용자 설명서에는 별도의 서명이 되어있지 않는데 이는 사용자 설명서가 본질적 동등성 승인서의 첨부문서로 여겨지기 때문이며 해당 승인서에 기재되어 있는 서명이 사용자 설명서에도 동일하게 적용이 된다.
만일 미 식약청이 목표기한을 10일 초과한 100일 이내에도 최종적인 결정을 내리지 못했을 경우 미 식약청은 제출자에게 서면으로 의사소통 권고문 (Missed MDUFA Communication)을 발행한다. 이 권고문은 미팅이나 원격 회의를 통한 협의가 필요한 미해결 사항이나 그 외에 최종 결정을 내리는데 방해가 되는 요소들과 그러한 사항들을 해결 할 수 있는 예상 일자가 기재되어 있다.
510(k) 신청의 추진 일정
미 식약청은 510(k)검토의 수행 목표로 의료기기 사용자 요금 개정법안 III(MDUFA III: Medical Device User Fee Amendments III)를 따른다. 아래의 도표는 510(k) 제출 절차의 주요 과정들에 대한 요약이다. 이 도표는 의료기기 사용자 요금 개정법안 III에 기재되어 있는 수행 목표와 절차 헌신의 서를 기반으로 작성 되었다. 더 상세한 내용은 MDUFA III의 수행 목표와 절차를 참고.
510(k) 검토간 의사소통 일정
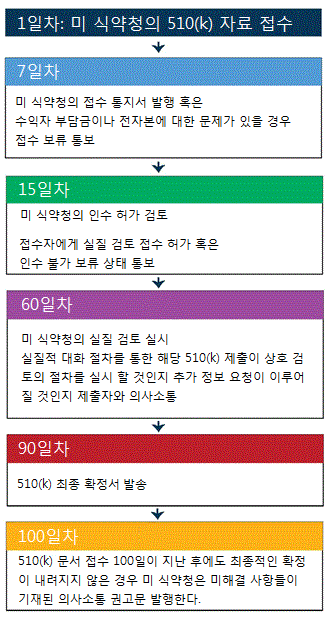
표기된 일자는 휴무일 제외 없이 역일로 계산 한다.
의사소통 일정은 2012년도 의료기기 사용자 요금 개정법안 III(MDUFA III: Medical Device User Fee Amendments III)에 기반한다.
위의 일정표 실제 절차가 간소화 된 것이다.
U.S. FDA 510(k) 지도 서비스에 대한 문의는 연락처의 정보로 문의 주시기 바랍니다.